publications
(*) denotes equal contribution.
2026
-
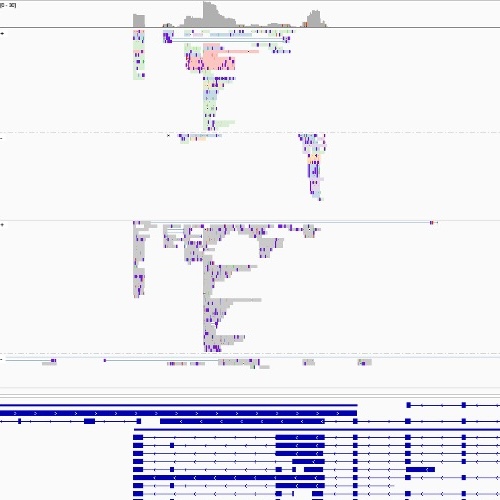
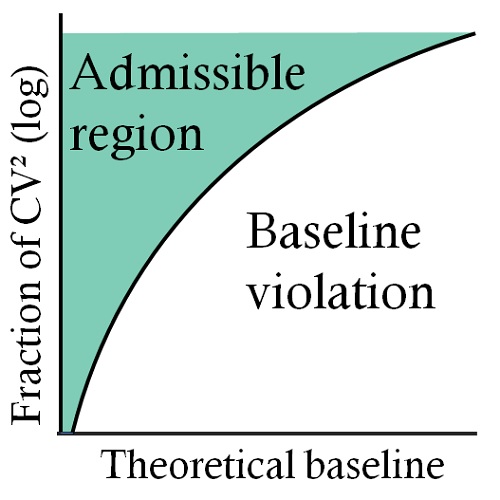
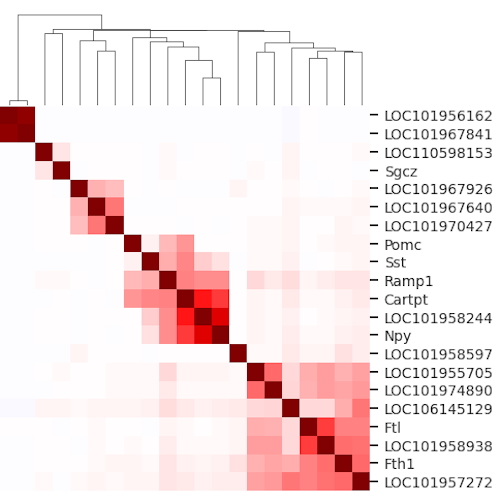 Empty drops in scRNA-seq uncover the surprising prevalence of sequestered neuropeptide mRNA and pervasive sequencing artifactsGennady Gorin, and Linda GoodmanbioRxiv, Feb 2026
Empty drops in scRNA-seq uncover the surprising prevalence of sequestered neuropeptide mRNA and pervasive sequencing artifactsGennady Gorin, and Linda GoodmanbioRxiv, Feb 2026The empty drops in single-cell sequencing experiments are an underexplored resource. As such, they present a substrate to ask questions orthogonal to standard single-cell sequencing workflows, calibrate statistical models using simple internal controls, and detect technical outliers which would be otherwise challenging to distinguish from real biology. In this case study, we report a relatively simple procedure to detect sequencing artifacts and make recommendations to reduce the risk of erroneous quantifications. In addition, we report the surprising abundance and co-expression of mRNA coding for neuropeptide-related genes in the empty drops, possibly reflecting underlying physiology.
-
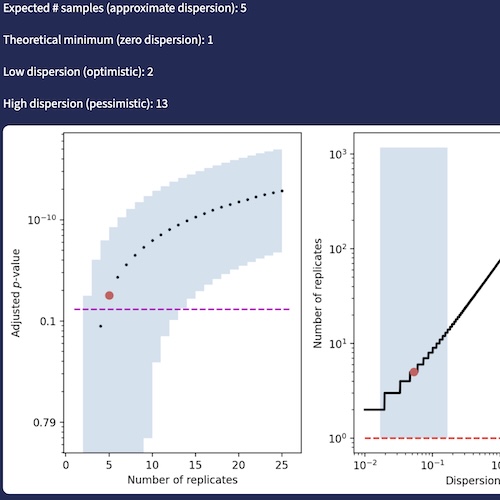 DEPower: approximate power analysis with DESeq2Gennady Gorin, Deek Guruge, and Linda GoodmanbioRxiv, Feb 2026
DEPower: approximate power analysis with DESeq2Gennady Gorin, Deek Guruge, and Linda GoodmanbioRxiv, Feb 2026Rigorous experimental design, including formal power analysis, is a cornerstone of reproducible RNA sequencing (RNA-seq) research. The design of RNA-seq experiments requires computing the minimum sample number required to identify an effect of a particular size at a predefined significance level. Ideally, the statistical test used for the analysis of experimental data should match the test used for sample size determination; however, few tools use the assumptions of the popular differential expression testing framework DESeq2, and most opt for simulation-based rather than analytical approaches. Grounded in the DESeq2 model framework, we derive sample size requirements for both single-cell and bulk RNA-seq experiments delivered as a web-based tool for power analysis, DEPower, available at https://poweranalysis-fb.streamlit.app/ that makes rigorous RNA-seq study design accessible to all researchers.
-
 Unexpected XIST Expression in Male Hearts Associates with DiseaseGennady Gorin, Natalie DeForest, and Linda GoodmanbioRxiv, Jan 2026
Unexpected XIST Expression in Male Hearts Associates with DiseaseGennady Gorin, Natalie DeForest, and Linda GoodmanbioRxiv, Jan 2026XIST is a long non-coding RNA that mediates the process of X chromosome inactivation in females, and has not been previously observed in males outside of developmental, pathological, and ectopic contexts. We report the unprecedented endogenous expression of XIST in 226 male heart samples across 15 single-cell RNA sequencing studies. The male expression of XIST is specific to a rare cell type typically annotated as neurons or glia. We investigate a variety of explanations and cross-reference the expression of XIST against age, sex, and pathology, finding signatures of a non-canonical inhibitory program, potentially mediated by a truncated transcript. Interestingly, the highest-powered datasets exhibit XIST upregulation in heart disease and comorbidity donors relative to healthier controls. Furthermore, in humans we identify a male-specific association of the XIST genomic locus with myocardial infarction (p = 1.3 × 10⁻⁴). Taken together, our findings suggest that the pathology of cardiomyopathy may involve uncharacterized, therapeutically actionable non-coding RNA pathways that operate through the cardiac nervous system, and underscore the importance of genomic investigations of the X chromosome.



2025
-
 Monod: model-based discovery and integration through fitting stochastic transcriptional dynamics to single-cell sequencing dataNature Methods, Nov 2025
Monod: model-based discovery and integration through fitting stochastic transcriptional dynamics to single-cell sequencing dataNature Methods, Nov 2025Single-cell RNA sequencing analysis centers on illuminating cell diversity and understanding the transcriptional mechanisms underlying cellular function. These datasets are large, noisy and complex. Current analyses prioritize noise removal and dimensionality reduction to tackle these challenges and extract biological insight. We propose an alternative, physical approach to leverage the stochasticity, size and multimodal nature of these data to explicitly distinguish their biological and technical facets while revealing the underlying regulatory processes. With the Python package Monod, we demonstrate how nascent and mature RNA counts, present in most published datasets, can be meaningfully ‘integrated’ under biophysical models of transcription. By using variation in these modalities, we can identify transcriptional modulation not discernible through changes in average gene expression, quantitatively compare mechanistic hypotheses of gene regulation, analyze transcriptional data from different technologies within a common framework and minimize the use of opaque or distortive normalization and transformation techniques.
-
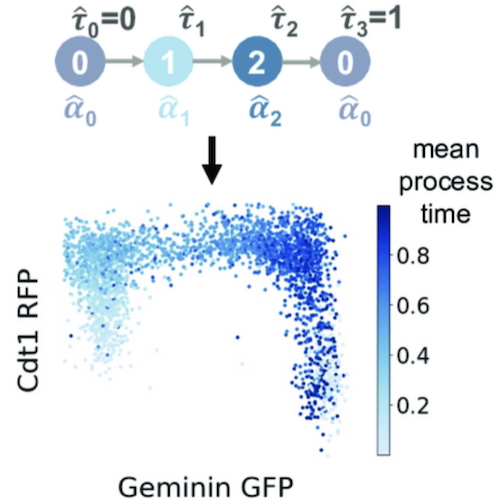 Trajectory inference from single-cell genomics data with a process time modelMeichen Fang, Gennady Gorin, and Lior PachterPLOS Computational Biology, Jan 2025
Trajectory inference from single-cell genomics data with a process time modelMeichen Fang, Gennady Gorin, and Lior PachterPLOS Computational Biology, Jan 2025Single-cell transcriptomics experiments provide gene expression snapshots of heterogeneous cell populations across cell states. These snapshots have been used to infer trajectories and dynamic information even without intensive, time-series data by ordering cells according to gene expression similarity. However, while single-cell snapshots sometimes offer valuable insights into dynamic processes, current methods for ordering cells are limited by descriptive notions of “pseudotime” that lack intrinsic physical meaning. Instead of pseudotime, we propose inference of “process time” via a principled modeling approach to formulating trajectories and inferring latent variables corresponding to timing of cells subject to a biophysical process. Our implementation of this approach, called Chronocell, provides a biophysical formulation of trajectories built on cell state transitions. The Chronocell model is identifiable, making parameter inference meaningful. Furthermore, Chronocell can interpolate between trajectory inference, when cell states lie on a continuum, and clustering, when cells cluster into discrete states. By using a variety of datasets ranging from cluster-like to continuous, we show that Chronocell enables us to assess the suitability of datasets and reveals distinct cellular distributions along process time that are consistent with biological process times. We also compare our parameter estimates of degradation rates to those derived from metabolic labeling datasets, thereby showcasing the biophysical utility of Chronocell. Nevertheless, based on performance characterization on simulations, we find that process time inference can be challenging, highlighting the importance of dataset quality and careful model assessment.


2024
-
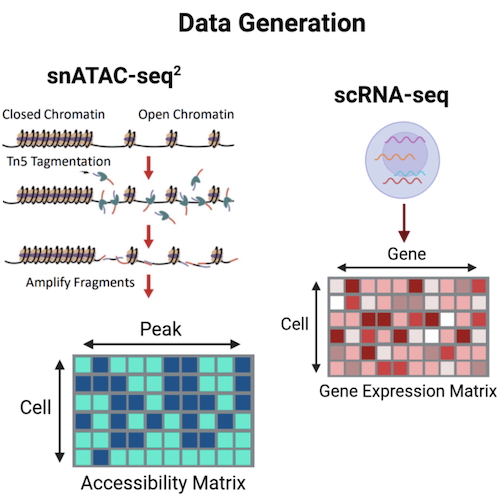 Biophysical model for joint analysis of chromatin and RNA sequencing dataCatherine Felce, Gennady Gorin, and Lior PachterPhys. Rev. E, Dec 2024
Biophysical model for joint analysis of chromatin and RNA sequencing dataCatherine Felce, Gennady Gorin, and Lior PachterPhys. Rev. E, Dec 2024The assay for transposase-accessible chromatin using sequencing (ATAC-seq) can be used to identify open chromatin regions, providing complementary information to RNA-seq which measures gene expression by sequencing. Single-cell multiome methods offer the possibility of measuring both modalities simultaneously in cells, raising the question of how to analyze them jointly, and also the extent to which the information they provide is better than unregistered data, where single-cell ATAC-seq and single-cell RNA-seq are performed on the same sample, but on different cells. We propose and motivate a biophysical model for chromatin dynamics and subsequent transcription that can be used to parametrize multiome data, and use it to assess the benefits of multiome data over unregistered single-cell RNA-seq and single-cell ATAC-seq. We also show that our model provides a biophysically grounded approach to the integration of chromatin accessibility data with other modalitie, and apply the model to single-cell ATAC-seq data.
-
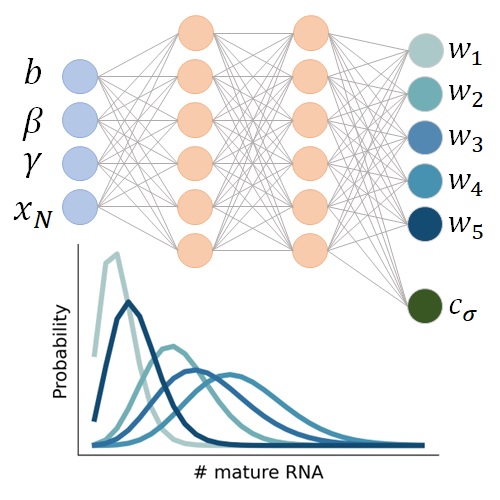 Spectral neural approximations for models of transcriptional dynamicsBiophysical Journal, Sep 2024
Spectral neural approximations for models of transcriptional dynamicsBiophysical Journal, Sep 2024The advent of high-throughput transcriptomics provides an opportunity to advance mechanistic understanding of transcriptional processes and their connections to cellular function at an unprecedented, genome-wide scale. These transcriptional systems, which involve discrete stochastic events, are naturally modeled using chemical master equations (CMEs), which can be solved for probability distributions to fit biophysical rates that govern system dynamics. While CME models have been used as standards in fluorescence transcriptomics for decades to analyze single-species RNA distributions, there are often no closed-form solutions to CMEs that model multiple species, such as nascent and mature RNA transcript counts. This has prevented the application of standard likelihood-based statistical methods for analyzing high-throughput, multi-species transcriptomic datasets using biophysical models. Inspired by recent work in machine learning to learn solutions to complex dynamical systems, we leverage neural networks and statistical understanding of system distributions to produce accurate approximations to a steady-state bivariate distribution for a model of the RNA life cycle that includes nascent and mature molecules. The steady-state distribution to this simple model has no closed-form solution and requires intensive numerical solving techniques: our approach reduces likelihood evaluation time by several orders of magnitude. We demonstrate two approaches, whereby solutions are approximated by 1) learning the weights of kernel distributions with constrained parameters or 2) learning both weights and scaling factors for parameters of kernel distributions. We show that our strategies, denoted by kernel weight regression and parameter-scaled kernel weight regression, respectively, enable broad exploration of parameter space and can be used in existing likelihood frameworks to infer transcriptional burst sizes, RNA splicing rates, and mRNA degradation rates from experimental transcriptomic data.
-
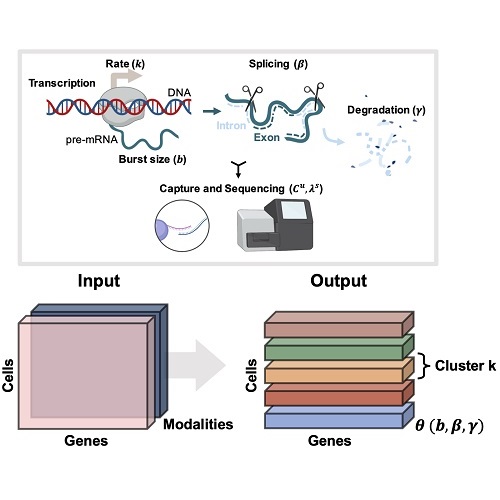 Biophysically interpretable inference of cell types from multimodal sequencing dataTara Chari, Gennady Gorin, and Lior PachterNature Computational Science, Sep 2024
Biophysically interpretable inference of cell types from multimodal sequencing dataTara Chari, Gennady Gorin, and Lior PachterNature Computational Science, Sep 2024Multimodal, single-cell genomics technologies enable simultaneous measurement of multiple facets of DNA and RNA processing in the cell. This creates opportunities for transcriptome-wide, mechanistic studies of cellular processing in heterogeneous cell populations, such as regulation of cell fate by transcriptional stochasticity or tumor proliferation through aberrant splicing dynamics. However, current methods for determining cell types or ‘clusters’ in multimodal data often rely on ad hoc approaches to balance or integrate measurements, and assumptions ignoring inherent properties of the data. To enable interpretable and consistent cell cluster determination, we present meK-means (mechanistic K-means) which integrates modalities through a unifying model of transcription to learn underlying, shared biophysical states. With meK-means we can cluster cells with nascent and mature mRNA measurements, utilizing the causal, physical relationships between these modalities. This identifies shared transcription dynamics across cells, which induce the observed molecule counts, and provides an alternative definition for ‘clusters’ through the governing parameters of cellular processes.
-
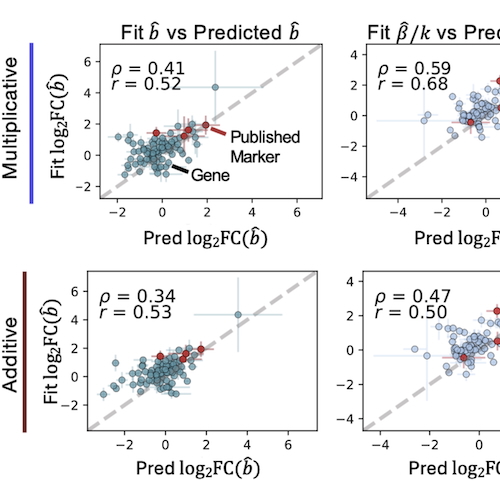 Stochastic Modeling of Biophysical Responses to PerturbationTara Chari, Gennady Gorin, and Lior PachterbioRxiv, Jul 2024
Stochastic Modeling of Biophysical Responses to PerturbationTara Chari, Gennady Gorin, and Lior PachterbioRxiv, Jul 2024Recent advances in high-throughput, multi-condition experiments allow for genome-wide investigation of how perturbations affect transcription and translation in the cell across multiple biological entities or modalities, from chromatin and mRNA information to protein production and spatial morphology. This presents an unprecedented opportunity to unravel how the processes of DNA and RNA regulation direct cell fate determination and disease response. Most methods designed for analyzing large-scale perturbation data focus on the observational outcomes, e.g., expression; however, many potential transcriptional mechanisms, such as transcriptional bursting or splicing dynamics, can underlie these complex and noisy observations. In this analysis, we demonstrate how a stochastic biophysical modeling approach to interpreting high-throughout perturbation data enables deeper investigation of the ’how’ behind such molecular measurements. Our approach takes advantage of modalities already present in data produced with current technologies, such as nascent and mature mRNA measurements, to illuminate transcriptional dynamics induced by perturbation, predict kinetic behaviors in new perturbation settings, and uncover novel populations of cells with distinct kinetic responses to perturbation.
-
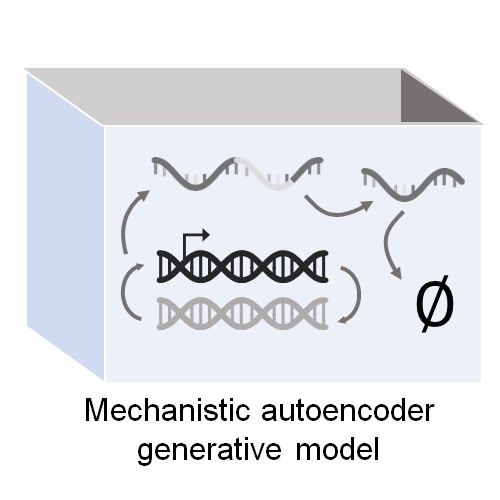 Biophysical modeling with variational autoencoders for bimodal, single-cell RNA sequencing dataNature Methods, Jul 2024
Biophysical modeling with variational autoencoders for bimodal, single-cell RNA sequencing dataNature Methods, Jul 2024Here we present biVI, which combines the variational autoencoder framework of scVI with biophysical models describing the transcription and splicing kinetics of RNA molecules. We demonstrate on simulated and experimental single-cell RNA sequencing data that biVI retains the variational autoencoder’s ability to capture cell type structure in a low-dimensional space while further enabling genome-wide exploration of the biophysical mechanisms, such as system burst sizes and degradation rates, that underlie observations.
-
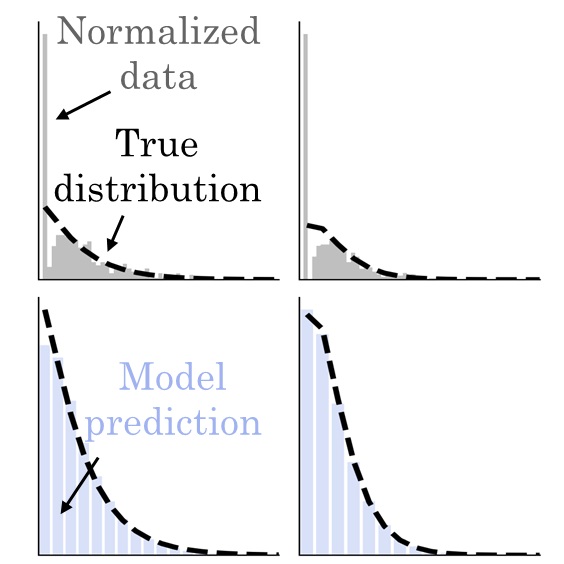 New and notable: Revisiting the ‘‘two cultures’’ through extrinsic noiseGennady Gorin, and Lior PachterBiophysical Journal, Jan 2024
New and notable: Revisiting the ‘‘two cultures’’ through extrinsic noiseGennady Gorin, and Lior PachterBiophysical Journal, Jan 2024A recent article by Grima and Esmenjaud draws attention to the unexpectedly complex effects of extrinsic noise on inference of transcriptional kinetics. We contrast the authors’ mechanistic approach with the descriptive, data-scientific methods used in single-cell RNA sequencing, and discuss broader philosophical connections to Leo Breiman’s "two cultures" of statistics.






2023
-
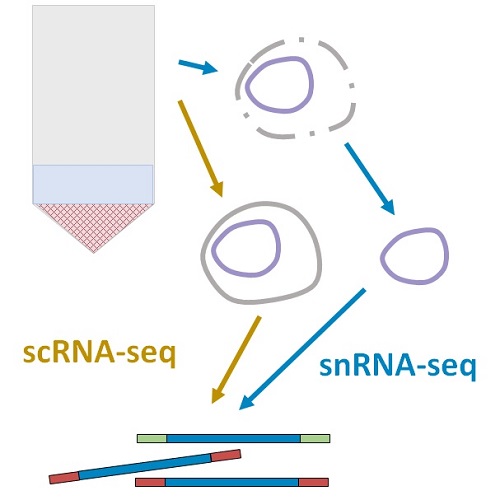 Assessing Markovian and Delay Models for Single-Nucleus RNA SequencingGennady Gorin, Shawn Yoshida, and Lior PachterBulletin of Mathematical Biology, Oct 2023
Assessing Markovian and Delay Models for Single-Nucleus RNA SequencingGennady Gorin, Shawn Yoshida, and Lior PachterBulletin of Mathematical Biology, Oct 2023The serial nature of reactions involved in the RNA life-cycle motivates the incorporation of delays in models of transcriptional dynamics. The models couple a transcriptional process to a fairly general set of delayed monomolecular reactions with no feedback. We provide numerical strategies for calculating the RNA copy number distributions induced by these models, and solve several systems with splicing, degradation, and catalysis. An analysis of single-cell and single-nucleus RNA sequencing data using these models reveals that the kinetics of nuclear export do not appear to require invocation of a non-Markovian waiting time.
-
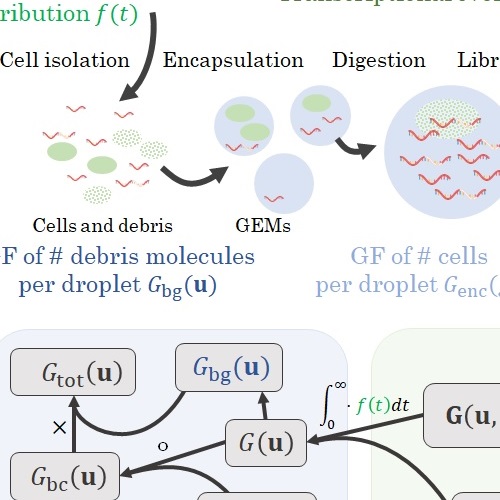 Studying stochastic systems biology of the cell with single-cell genomics dataGennady Gorin, John J. Vastola, and Lior PachterCell Systems, Oct 2023
Studying stochastic systems biology of the cell with single-cell genomics dataGennady Gorin, John J. Vastola, and Lior PachterCell Systems, Oct 2023Recent experimental developments in genome-wide RNA quantification hold considerable promise for systems biology. However, rigorously probing the biology of living cells requires a unified mathematical framework that accounts for single-molecule biological stochasticity in the context of technical variation associated with genomics assays. We review models for a variety of RNA transcription processes, as well as the encapsulation and library construction steps of microfluidics-based single-cell RNA sequencing, and present a framework to integrate these phenomena by the manipulation of generating functions. Finally, we use simulated scenarios and biological data to illustrate the implications and applications of the approach.
-
 Stochastic foundations for single-cell RNA sequencingGennady GorinMay 2023
Stochastic foundations for single-cell RNA sequencingGennady GorinMay 2023Single-cell RNA sequencing, which quantifies cell transcriptomes, has seen widespread adoption, accompanied by proliferation of analysis methods. However, there has been relatively little systematic investigation of its best practices and their underlying assumptions, leading to challenges and discrepancies in interpretation. I present a set of generic, principled strategies for modeling the biological and technical components of sequencing experiments and use case studies to motivate their application to sequencing data.
-
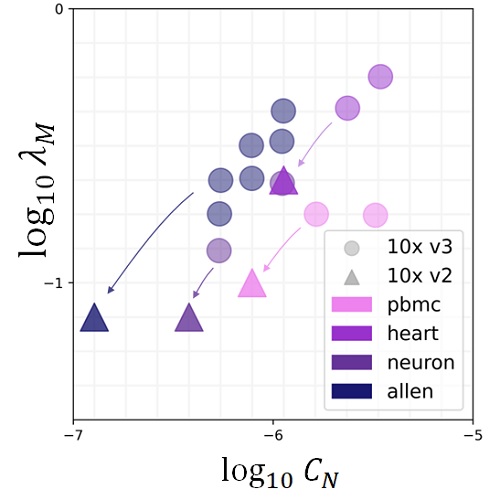 Length biases in single-cell RNA sequencing of pre-mRNAGennady Gorin, and Lior PachterBiophysical Reports, Mar 2023
Length biases in single-cell RNA sequencing of pre-mRNAGennady Gorin, and Lior PachterBiophysical Reports, Mar 2023Single-cell RNA sequencing data can be modeled using Markov chains to yield genome-wide insights into transcriptional physics. However, quantitative inference with such data requires careful assessment of noise sources. We find that long pre-mRNA transcripts are over-represented in sequencing data. To explain this trend, we propose a length-based model of capture bias, which may produce false-positive observations. We solve this model and use it to find concordant parameter trends as well as systematic, mechanistically interpretable technical and biological differences in paired data sets.
-
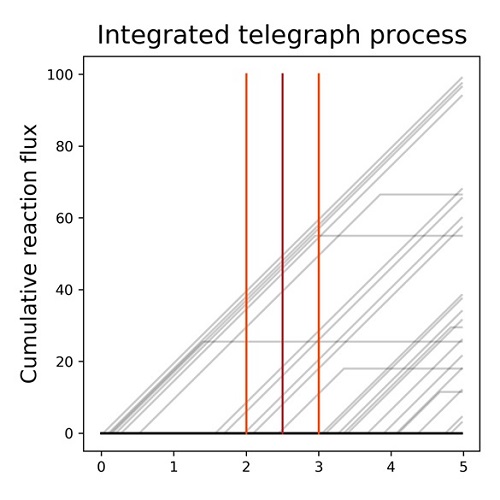 The telegraph process is not a subordinatorGennady Gorin, and Lior PachterbioRxiv, Jan 2023
The telegraph process is not a subordinatorGennady Gorin, and Lior PachterbioRxiv, Jan 2023Investigations of transcriptional models by Amrhein et al. outline a strategy for connecting steady-state distributions to process dynamics. We clarify its limitations: the strategy holds for a very narrow class of processes, which excludes an example given by the authors.





2022
-
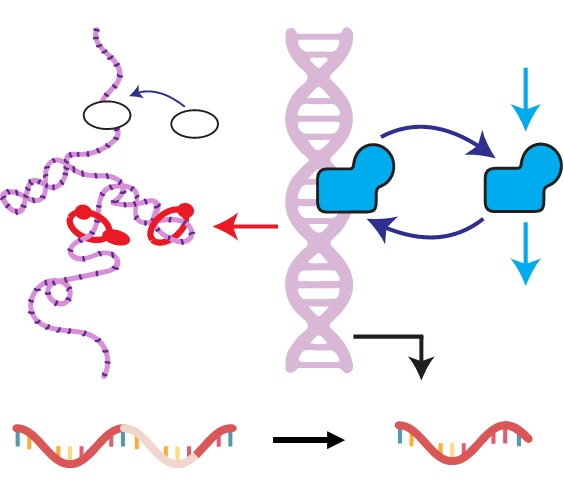 Interpretable and tractable models of transcriptional noise for the rational design of single-molecule quantification experimentsNature Communications, Dec 2022
Interpretable and tractable models of transcriptional noise for the rational design of single-molecule quantification experimentsNature Communications, Dec 2022Abstract The question of how cell-to-cell differences in transcription rate affect RNA count distributions is fundamental for understanding biological processes underlying transcription. Answering this question requires quantitative models that are both interpretable (describing concrete biophysical phenomena) and tractable (amenable to mathematical analysis). This enables the identification of experiments which best discriminate between competing hypotheses. As a proof of principle, we introduce a simple but flexible class of models involving a continuous stochastic transcription rate driving a discrete RNA transcription and splicing process, and compare and contrast two biologically plausible hypotheses about transcription rate variation. One assumes variation is due to DNA experiencing mechanical strain, while the other assumes it is due to regulator number fluctuations. We introduce a framework for numerically and analytically studying such models, and apply Bayesian model selection to identify candidate genes that show signatures of each model in single-cell transcriptomic data from mouse glutamatergic neurons.
-
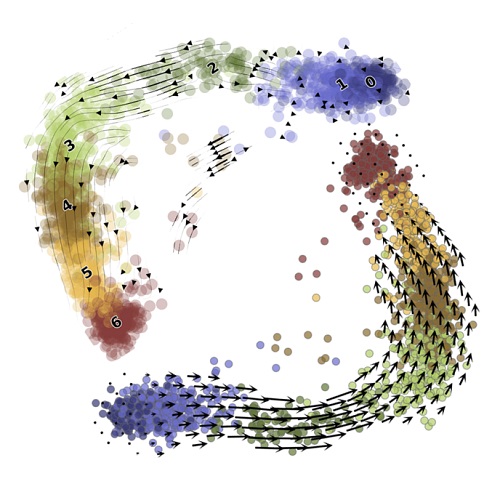 RNA velocity unraveledPLOS Computational Biology, Sep 2022
RNA velocity unraveledPLOS Computational Biology, Sep 2022We perform a thorough analysis of RNA velocity methods, with a view towards understanding the suitability of the various assumptions underlying popular implementations. In addition to providing a self-contained exposition of the underlying mathematics, we undertake simulations and perform controlled experiments on biological datasets to assess workflow sensitivity to parameter choices and underlying biology. Finally, we argue for a more rigorous approach to RNA velocity, and present a framework for Markovian analysis that points to directions for improvement and mitigation of current problems.
-
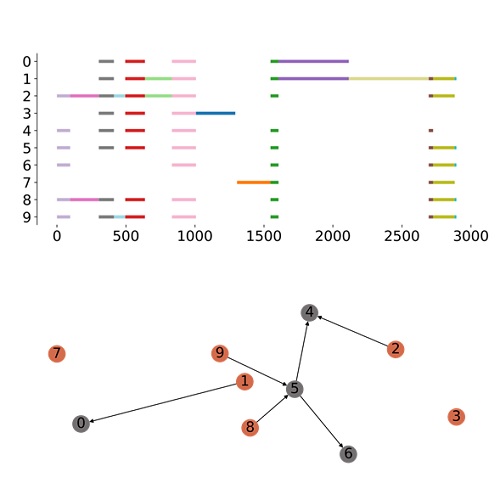 Modeling bursty transcription and splicing with the chemical master equationGennady Gorin, and Lior PachterBiophysical Journal, Feb 2022
Modeling bursty transcription and splicing with the chemical master equationGennady Gorin, and Lior PachterBiophysical Journal, Feb 2022Splicing cascades that alter gene products post-transcriptionally also affect expression dynamics. We study a class of processes and associated distributions that emerge from models of bursty promoters coupled to directed acyclic graphs of splicing. These solutions provide full time-dependent joint distributions for an arbitrary number of species with general noise behaviors and transient phenomena, offering qualitative and quantitative insights about how splicing can regulate expression dynamics. Finally, we derive a set of quantitative constraints on the minimum complexity necessary to reproduce gene coexpression patterns using synchronized burst models. We validate these findings by analyzing long-read sequencing data, where we find evidence of expression patterns largely consistent with these constraints.



2021
-
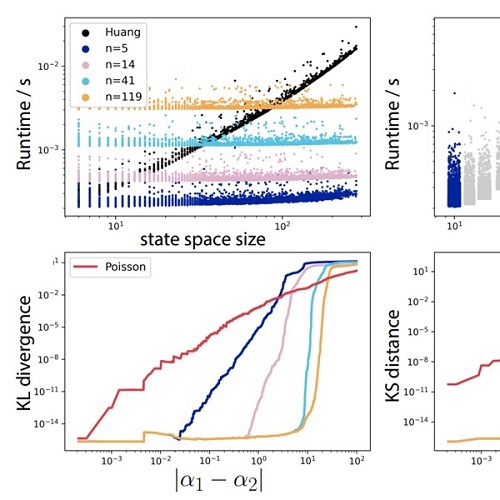 Analytic solution of chemical master equations involving gene switching. I: Representation theory and diagrammatic approach to exact solutionarXiv, Mar 2021
Analytic solution of chemical master equations involving gene switching. I: Representation theory and diagrammatic approach to exact solutionarXiv, Mar 2021The chemical master equation (CME), which describes the discrete and stochastic molecule number dynamics associated with biological processes like transcription, is difficult to solve analytically. It is particularly hard to solve for models involving bursting/gene switching, a biological feature that tends to produce heavy-tailed single cell RNA counts distributions. In this paper, we present a novel method for computing exact and analytic solutions to the CME in such cases, and use these results to explore approximate solutions valid in different parameter regimes, and to compute observables of interest. Our method leverages tools inspired by quantum mechanics, including ladder operators and Feynman-like diagrams, and establishes close formal parallels between the dynamics of bursty transcription, and the dynamics of bosons interacting with a single fermion. We focus on two problems: (i) the chemical birth-death process coupled to a switching gene/the telegraph model, and (ii) a model of transcription and multistep splicing involving a switching gene and an arbitrary number of downstream splicing steps. We work out many special cases, and exhaustively explore the special functionology associated with these problems. This is Part I in a two-part series of papers; in Part II, we explore an alternative solution approach that is more useful for numerically solving these problems, and apply it to parameter inference on simulated RNA counts data.

2020
-
 Intrinsic and extrinsic noise are distinguishable in a synthesis – export – degradation model of mRNA productionGennady Gorin, and Lior PachterbioRxiv, Sep 2020
Intrinsic and extrinsic noise are distinguishable in a synthesis – export – degradation model of mRNA productionGennady Gorin, and Lior PachterbioRxiv, Sep 2020Intrinsic and extrinsic noise sources in gene expression, originating respectively from transcriptional stochasticity and from differences between cells, complicate the determination of transcriptional models. In particularly degenerate cases, the two noise sources are altogether impossible to distinguish. However, the incorporation of downstream processing, such as the mRNA splicing and export implicated in gene expression buffering, recovers the ability to identify the relevant source of noise. We report analytical copy-number distributions, discuss the noise sources’ qualitative effects on lower moments, and provide simulation routines for both models.
-
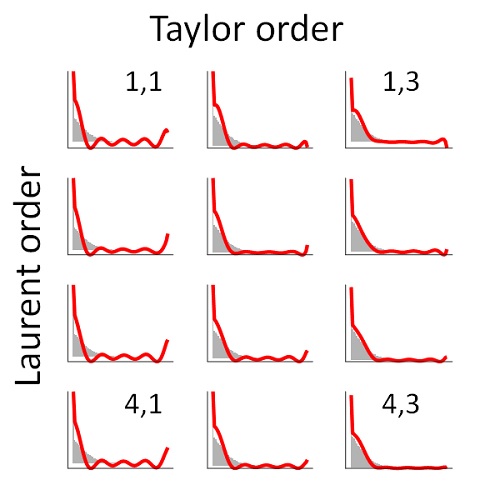 Special function methods for bursty models of transcriptionGennady Gorin, and Lior PachterPhysical Review E, Aug 2020
Special function methods for bursty models of transcriptionGennady Gorin, and Lior PachterPhysical Review E, Aug 2020We explore a Markov model used in the analysis of gene expression, involving the bursty production of pre-mRNA, its conversion to mature mRNA, and its consequent degradation. We demonstrate that the integration used to compute the solution of the stochastic system can be approximated by the evaluation of special functions. Furthermore, the form of the special function solution generalizes to a broader class of burst distributions. In light of the broader goal of biophysical parameter inference from transcriptomics data, we apply the method to simulated data, demonstrating effective control of precision and runtime. Finally, we propose and validate a non-Bayesian approach for parameter estimation based on the characteristic function of the target joint distribution of pre-mRNA and mRNA.
-
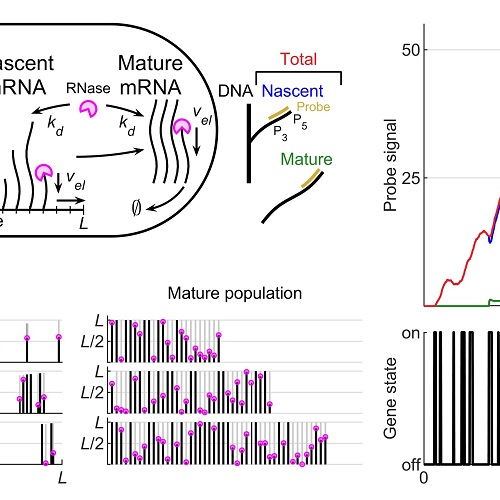 Stochastic simulation and statistical inference platform for visualization and estimation of transcriptional kineticsPLOS ONE, Mar 2020
Stochastic simulation and statistical inference platform for visualization and estimation of transcriptional kineticsPLOS ONE, Mar 2020Recent advances in single-molecule fluorescent imaging have enabled quantitative measurements of transcription at a single gene copy, yet an accurate understanding of transcriptional kinetics is still lacking due to the difficulty of solving detailed biophysical models. Here we introduce a stochastic simulation and statistical inference platform for modeling detailed transcriptional kinetics in prokaryotic systems, which has not been solved analytically. The model includes stochastic two-state gene activation, mRNA synthesis initiation and stepwise elongation, release to the cytoplasm, and stepwise co-transcriptional degradation. Using the Gillespie algorithm, the platform simulates nascent and mature mRNA kinetics of a single gene copy and predicts fluorescent signals measurable by time-lapse single-cell mRNA imaging, for different experimental conditions. To approach the inverse problem of estimating the kinetic parameters of the model from experimental data, we develop a heuristic optimization method based on the genetic algorithm and the empirical distribution of mRNA generated by simulation. As a demonstration, we show that the optimization algorithm can successfully recover the transcriptional kinetics of simulated and experimental gene expression data. The platform is available as a MATLAB software package at https://data.caltech.edu/records/1287.
-
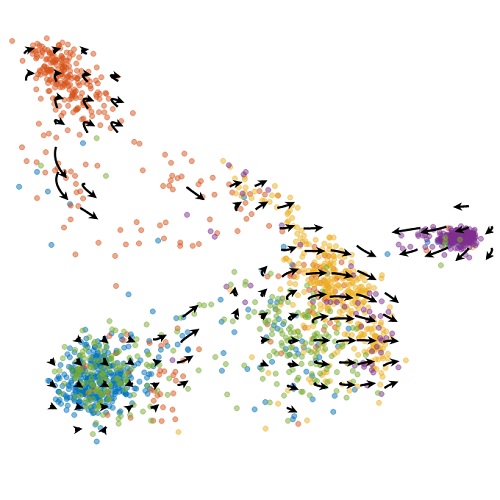 Protein velocity and acceleration from single-cell multiomics experimentsGennady Gorin, Valentine Svensson, and Lior PachterGenome Biology, Feb 2020
Protein velocity and acceleration from single-cell multiomics experimentsGennady Gorin, Valentine Svensson, and Lior PachterGenome Biology, Feb 2020The simultaneous quantification of protein and RNA makes possible the inference of past, present, and future cell states from single experimental snapshots. To enable such temporal analysis from multimodal single-cell experiments, we introduce an extension of the RNA velocity method that leverages estimates of unprocessed transcript and protein abundances to extrapolate cell states. We apply the model to six datasets and demonstrate consistency among cell landscapes and phase portraits. The analysis software is available as the protaccel Python package.



